A guide to metadata for samples and differential expression analyses
B. Ogan Mancarci
Michael Smith Laboratories, University of British Columbia, Vancouver, CanadaSource:
vignettes/metadata.Rmd
metadata.RmdIntroduction
The data in Gemma are manually annotated by curators with terms, often using an ontology term on both dataset and sample level. In Gemma.R three primary functions allow access to these annotations for a given dataset.
get_dataset_annotations: This function returns annotations associated with a dataset. These try to serve as tags describing the dataset as a whole and they characteristics that samples within the datasets have while also including some additional terms.get_dataset_samples: This function returns samples and associated annotations related to their experimental groups for an experimentget_dataset_differential_expression_analyses: This function returns information about differential expression analyses automatically performed by Gemma for a given experiment. Each row of the output is a contrast where a specific property or an interaction of properties are described.
In the examples below we will be referring to GSE48962 experiment, where striatum and cerebral cortex samples from control mice and mice belonging to a Huntington model (R6/2) were taken from 8 week and 12 week old mice.
Dataset tags
Terms returned via get_dataset_annotations are tags used
to describe a dataset in general terms.
get_dataset_annotations('GSE48962') %>%
gemma_kable| class.name | class.URI | term.name | term.URI | object.class |
|---|---|---|---|---|
| assay | http://purl.obolibrary…/OBI_0000070 | bulk RNA-seq assay | http://purl.obolibrary…/OBI_0003090 | ExperimentTag |
| organism part | http://www.ebi…/EFO_0000635 | striatum | http://purl.obolibrary…/UBERON_0002435 | FactorValue |
| organism part | http://www.ebi…/EFO_0000635 | cerebral cortex | http://purl.obolibrary…/UBERON_0000956 | FactorValue |
| genotype | http://www.ebi…/EFO_0000513 | R6/2 | http://gemma.msl…/TGEMO_00178 | FactorValue |
| developmental stage | http://www.ebi…/EFO_0000399 | prime adult stage | http://purl.obolibrary…/UBERON_0018241 | FactorValue |
These tags come as a class/term pairs and inherit any terms that is assigned to any of the samples. Therefore we can see all chemicals and cell types used in the experiment.
Factor values
Samples and differential expression contrasts in Gemma are annotated with factor values. These values contain statements that describe these samples and which samples belong to which experimental in a differential expression analysis respectively.
Sample factor values
In gemma.R these values are stored in nested data.tables
and can be found by accessing the relevant columns of the outputs.
Annotations for samples can be accessed using
get_dataset_samples. sample.factorValues
column contains the relevant information
samples <- get_dataset_samples('GSE48962')
samples$sample.factorValues[[
which(samples$sample.name == "TSM490")
]] %>%
gemma_kable()| category | category.URI | value | value.URI | predicate | predicate.URI | object | object.URI | summary | ID | factor.ID | factor.category | factor.category.URI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| organism part | http://www.ebi…/EFO_0000635 | striatum | http://purl.obolibrary…/UBERON_0002435 | NA | NA | NA | NA | striatum | 120172 | 20540 | organism part | http://www.ebi…/EFO_0000635 |
| developmental stage | http://www.ebi…/EFO_0000399 | prime adult stage | http://purl.obolibrary…/UBERON_0018241 | has developmental stage | http://gemma.msl…/TGEMO_00168 | 12 week | NA | 12 week prime adult stage | 120179 | 20543 | developmental stage | http://www.ebi…/EFO_0000399 |
| block | http://www.ebi…/EFO_0005067 | Device=HWUSI-EAS1563_0073_F… | NA | NA | NA | NA | NA | Device=HWUSI-EAS1563_0073_F… | 163476 | 32643 | block | http://www.ebi…/EFO_0005067 |
| genotype | http://www.ebi…/EFO_0000513 | R6/2 | http://gemma.msl…/TGEMO_00178 | NA | NA | NA | NA | R6/2 | 120175 | 20541 | strain | http://www.ebi…/EFO_0005135 |
The example above shows a single factor value object for one sample.
The rows of this data.table are statements that belong to a
factor value. Below each column of this nested table is described. If a
given field is filled by an ontology term, the corresponding URI column
will contain the ontology URI for the field.
-
category/category.URI: Category of the individual statement, such as treatment, phenotype or strain -
value/value.URI: The subject of the statement. -
predicate/predicate.URI: When a subject alone is not enough to describe all details, a statement can contain a predicate and an object. The predicate describes the relationship between the subject of the statement and the object. In the example above, these are used to denote properties of the human HTT in the mouse models -
object/object.URI: The object of a statement is a property further describing it’s value. In this example these describe the properties of the HTT gene in the mouse model, namely that it has CAG repeats and it is overexpressed. If the value was a drug this could be dosage or timepoint. -
summary: A plain text summary of the factorValue. Different statements will have the same summary if they are part of the same factor value -
ID: An integer identifier for the specific factor value. In the example above, the genotype of the mouse is defined as a single factor value made up of two statements stating the HTT gene has CAG repeats and that it is overexpressed. This factor value has the ID of 120175 which is shared by both rows containing the statements describing it. This ID will repeat for every other patient that has the same genotype or differential expression results using that factor as a part of their contrast. For instance we can see which samples that was subjected to this condition using this ID instead of trying to match the other columns describing the statements
id <- samples$sample.factorValues[[
which(samples$sample.name == "TSM490")
]] %>% filter(value == "R6/2") %>% {.$ID} %>% unique
# count how many patients has this phenotype
samples$sample.factorValues %>% sapply(\(x){
id %in% x$ID
}) %>% sum## [1] 12-
factor.ID: An integer identifier for the factor. A factor holds specific factor values. For the example above whether or not the mouse is a wild type mouse or if it has a wild type genotype is stored under the id
We can use this to fetch all distinct genotypes
id <- samples$sample.factorValues[[
which(samples$sample.name == "TSM490")
]] %>%
filter(value == "R6/2") %>% {.$factor.ID} %>% unique
samples$sample.factorValues %>% lapply(\(x){
x %>% filter(factor.ID == id) %>% {.$summary}
}) %>% unlist %>% unique## [1] "C57BL/6 x CBA has role control" "R6/2"This shows us the dataset has control mice and Huntington Disease
model mice.. This ID can be used to match the factor between samples and
between samples and differential expression experiments -
factor.category/factor.category.URI: The
category of the whole factor. Usually this is the same with the
category of the statements making up the factor value.
However in cases like the example above, where the value describes a
treatment while the factor overall represents a phenotype, they can
differ.
gemma.R includes a convenience function to create a simplified design matrix out of these factor values for a given experiment. This will unpack the nested data.frames and provide a more human readable output, giving each available factor it’s own column.
design <- make_design(samples)
design[,-1] %>% head %>% # first column is just a copy of the original factor values
gemma_kable()| organism part | developmental stage | block | strain | |
|---|---|---|---|---|
| ESW176 | striatum | 8 week prime adult stage | Device=HWUSI-EAS1563_0071_F… | C57BL/6 x CBA has role control |
| TCW9469 | cerebral cortex | 12 week prime adult stage | Device=HWUSI-EAS1563_0053:L… | C57BL/6 x CBA has role control |
| ECM175 | cerebral cortex | 8 week prime adult stage | Device=HWUSI-EAS1563_0071_F… | R6/2 |
| ESW183 | striatum | 8 week prime adult stage | Device=HWUSI-EAS1563_0072_F… | C57BL/6 x CBA has role control |
| ECW178 | cerebral cortex | 8 week prime adult stage | Device=HWUSI-EAS1563_0071_F… | C57BL/6 x CBA has role control |
| TSW479 | striatum | 12 week prime adult stage | Device=HWUSI-EAS1563_0073_F… | C57BL/6 x CBA has role control |
Using this output, here we look at the sample sizes for different experimental groups.
design %>%
group_by(`organism part`,`developmental stage`,strain) %>%
summarize(n= n()) %>%
arrange(desc(n)) %>%
gemma_kable()## `summarise()` has regrouped the output.
## ℹ Summaries were computed grouped by organism part, developmental stage, and
## strain.
## ℹ Output is grouped by organism part and developmental stage.
## ℹ Use `summarise(.groups = "drop_last")` to silence this message.
## ℹ Use `summarise(.by = c(organism part, developmental stage, strain))` for
## per-operation grouping (`?dplyr::dplyr_by`) instead.| organism part | developmental stage | strain | n |
|---|---|---|---|
| cerebral cortex | 12 week prime adult stage | C57BL/6 x CBA has role control | 3 |
| cerebral cortex | 12 week prime adult stage | R6/2 | 3 |
| cerebral cortex | 8 week prime adult stage | C57BL/6 x CBA has role control | 3 |
| cerebral cortex | 8 week prime adult stage | R6/2 | 3 |
| striatum | 12 week prime adult stage | C57BL/6 x CBA has role control | 3 |
| striatum | 12 week prime adult stage | R6/2 | 3 |
| striatum | 8 week prime adult stage | C57BL/6 x CBA has role control | 3 |
| striatum | 8 week prime adult stage | R6/2 | 3 |
Differential expression analysis factor values
For most experiments it contains, Gemma performs automated differential expression analyses. The kinds of analyses that will be performed is informed by the factor values belonging to the samples.
# removing columns containing factor values and URIs for brevity
remove_columns <- c('baseline.factors','experimental.factors','subsetFactor','factor.category.URI')
dea <- get_dataset_differential_expression_analyses("GSE48962")
dea[,.SD,.SDcols = !remove_columns] %>%
gemma_kable()| result.ID | contrast.ID | experiment.ID | factor.category | factor.ID | isSubset | probes.analyzed | genes.analyzed |
|---|---|---|---|---|---|---|---|
| 492853 | 120175_120178 | 8972 | developmental stage,strain | 20541,20543 | TRUE | 20621 | 20623 |
| 492852 | 120175 | 8972 | strain | 20541 | TRUE | 20621 | 20623 |
| 492851 | 120178 | 8972 | developmental stage | 20543 | TRUE | 20621 | 20623 |
| 492856 | 120175_120178 | 8972 | developmental stage,strain | 20541,20543 | TRUE | 18969 | 18972 |
| 492855 | 120175 | 8972 | strain | 20541 | TRUE | 18968 | 18971 |
| 492854 | 120178 | 8972 | developmental stage | 20543 | TRUE | 18968 | 18971 |
The example above shows the differential expression analyses results.
Each row of this data.table represents a differential expression
contrast connected to a fold change and a p value in the output of
get_differential_expression_values function. If we look at
the contrast.ID we will see the factor value identifiers
returned in the ID column of our
sample.factorValues. These represent which factor value is
used as the experimental factor. Note that some rows will have two IDs
appended together. These represent the interaction effects of multiple
factors. For simplicity, we will start from a contrast without an
interaction.
contrast <- dea %>%
filter(
factor.category == "strain" &
subsetFactor %>% map_chr('value') %>% {.=='cerebral cortex'} # we will talk about subsets in a moment
)
# removing URIs for brevity
uri_columns = c('category.URI',
'object.URI',
'value.URI',
'predicate.URI',
'factor.category.URI')
contrast$baseline.factors[[1]][,.SD,.SDcols = !uri_columns] %>%
gemma_kable()| category | value | predicate | object | summary | ID | factor.ID | factor.category |
|---|---|---|---|---|---|---|---|
| genotype | C57BL/6 x CBA | has role | control | C57BL/6 x CBA has role control | 120174 | 20541 | strain |
contrast$experimental.factors[[1]][,.SD,.SDcols = !uri_columns] %>%
gemma_kable()| category | value | predicate | object | summary | ID | factor.ID | factor.category |
|---|---|---|---|---|---|---|---|
| genotype | R6/2 | NA | NA | R6/2 | 120175 | 20541 | strain |
Here, we can see the baseline is the wild type mouse, being compared to the Huntington Disease models
If we examine a factor with interaction, both baseline and experimental factor value columns will contain two factor values.
contrast <- dea %>%
filter(
factor.category == "developmental stage,strain" &
subsetFactor %>% map_chr('value') %>% {.=='cerebral cortex'} # we're almost there!
)
contrast$baseline.factors[[1]][,.SD,.SDcols = !uri_columns] %>%
gemma_kable()| category | value | predicate | object | summary | ID | factor.ID | factor.category |
|---|---|---|---|---|---|---|---|
| genotype | C57BL/6 x CBA | has role | control | C57BL/6 x CBA has role control | 120174 | 20541 | strain |
| developmental stage | prime adult stage | has developmental stage | 12 week | 12 week prime adult stage | 120179 | 20543 | developmental stage |
contrast$experimental.factors[[1]][,.SD,.SDcols = !uri_columns] %>%
gemma_kable()| category | value | predicate | object | summary | ID | factor.ID | factor.category |
|---|---|---|---|---|---|---|---|
| genotype | R6/2 | NA | NA | R6/2 | 120175 | 20541 | strain |
| developmental stage | prime adult stage | has developmental stage | 8 week | 8 week prime adult stage | 120178 | 20543 | developmental stage |
A third place that can contain factorValues is the
subsetFactor. Certain differential expression analyses
exclude certain samples based on a given factor. In this example we can
see that this analysis were only performed on samples from the cerebral
cortex.
contrast$subsetFactor[[1]][,.SD,.SDcols = !uri_columns] %>%
gemma_kable()| category | value | predicate | object | summary | ID | factor.ID | factor.category |
|---|---|---|---|---|---|---|---|
| organism part | cerebral cortex | NA | NA | cerebral cortex | 120173 | 20540 | organism part |
The ids of the factor values included in
baseline.factors and experimental.factors
along with subsetFactor can be used to determine which
samples represent a given contrast. For convenience,
get_dataset_object function which is used to compile
metadata and expression data of an experiment in a single object,
includes resultSets and contrasts argument
which will return the data already composed of samples representing a
particular contrast.
obj <- get_dataset_object("GSE48962",resultSets = contrast$result.ID,contrasts = contrast$contrast.ID,type = 'list')
obj[[1]]$design[,-1] %>%
head %>% gemma_kable()| organism part | developmental stage | block | strain | |
|---|---|---|---|---|
| TCW9469 | cerebral cortex | 12 week prime adult stage | Device=HWUSI-EAS1563_0053:L… | C57BL/6 x CBA has role control |
| ECM175 | cerebral cortex | 8 week prime adult stage | Device=HWUSI-EAS1563_0071_F… | R6/2 |
| ECW178 | cerebral cortex | 8 week prime adult stage | Device=HWUSI-EAS1563_0071_F… | C57BL/6 x CBA has role control |
| TCW9451 | cerebral cortex | 12 week prime adult stage | Device=HWI-EAS413_0047:Lane=2 | C57BL/6 x CBA has role control |
| TCW9457 | cerebral cortex | 12 week prime adult stage | Device=HWUSI-EAS1563_0053:L… | C57BL/6 x CBA has role control |
| TCM9450 | cerebral cortex | 12 week prime adult stage | Device=HWI-EAS413_0047:Lane=1 | R6/2 |
We suggested that the contrast.ID of a contrast also
corresponded to a column in the differential expression results,
acquired by get_differential_expression_values. We can use
what we have learned to take a look at the expression of genes at the
top of the phenotype, treatment interaction. Each result.ID returns its
separate table when accessing differential expression values.
dif_vals <- get_differential_expression_values('GSE48962')
dif_vals[[as.character(contrast$result.ID)]] %>% head %>%
gemma_kable()| Probe | NCBIid | gene_ensembl_id | GeneSymbol | GeneName | pvalue | corrected_pvalue | rank | contrast_120175_120178_coefficient | contrast_120175_120178_log2fc | contrast_120175_120178_tstat | contrast_120175_120178_pvalue |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 100502959 | 100502959 | AV051173 | expressed sequence AV051173 | 1.60e-06 | 0.0163 | 9.70e-05 | 3.7586 | 3.7586 | 12.4896 | 1.60e-06 | |
| 67993 | 67993 | ENSMUSG00000024228 | Nudt12 | nudix hydrolase 12 | 1.10e-06 | 0.0163 | 4.85e-05 | -1.1088 | -1.1088 | -13.0595 | 1.10e-06 |
| 108096 | 108096 | ENSMUSG00000063975 | Slco1a5 | solute carrier organic anio… | 1.00e-04 | 0.2082 | 6.00e-04 | -3.6495 | -3.6495 | -6.7998 | 1.00e-04 |
| 101883 | 101883 | ENSMUSG00000036826 | Igflr1 | IGF-like family receptor 1 | 1.00e-04 | 0.2082 | 6.00e-04 | 2.3595 | 2.3595 | 6.7859 | 1.00e-04 |
| 51795 | 51795 | ENSMUSG00000090084 | Srpx | sushi-repeat-containing pro… | 1.00e-04 | 0.2082 | 5.00e-04 | -4.4992 | -4.4992 | -6.8665 | 1.00e-04 |
| 16969 | 16969 | ENSMUSG00000035011 | Zbtb7a | zinc finger and BTB domain … | 7.81e-05 | 0.2082 | 2.00e-04 | 1.1581 | 1.1581 | 7.3738 | 7.81e-05 |
To get the top genes found associated with this interaction we access
the columns with the correct contrast.ID.
# getting the top 10 genes
top_genes <- dif_vals[[as.character(contrast$result.ID)]] %>%
arrange(across(paste0('contrast_',contrast$contrast.ID,'_pvalue'))) %>%
filter(GeneSymbol!='' | grepl("|",GeneSymbol,fixed = TRUE)) %>% # remove blank genes or probes with multiple genes
{.[1:10,]}
top_genes %>% select(Probe,NCBIid,GeneSymbol) %>%
gemma_kable()| Probe | NCBIid | GeneSymbol |
|---|---|---|
| 67993 | 67993 | Nudt12 |
| 100502959 | 100502959 | AV051173 |
| 12153 | 12153 | Bmp1 |
| 58212 | 58212 | Srrm3 |
| 16969 | 16969 | Zbtb7a |
| 19732 | 19732 | Rgl2 |
| 108096 | 108096 | Slco1a5 |
| 101883 | 101883 | Igflr1 |
| 51795 | 51795 | Srpx |
| 108168973 | 108168973 | Gm12828 |
We can then use the expression data returned by
get_dataset_object to examine the expression values for
these genes.
exp_subset<- obj[[1]]$exp %>%
filter(Probe %in% top_genes$Probe)
genes <- top_genes$GeneSymbol
# ordering design file
design <- obj[[1]]$design %>% arrange(strain,`developmental stage`)
# shorten the resistance label a bit
design$strain[grepl('R6/2',design$strain)] = "Huntington Model"
exp_subset[,.SD,.SDcols = rownames(design)] %>% t %>% scale %>% t %>%
pheatmap(cluster_rows = FALSE,cluster_cols = FALSE,labels_row = genes,
annotation_col =design %>% select(strain,`developmental stage`))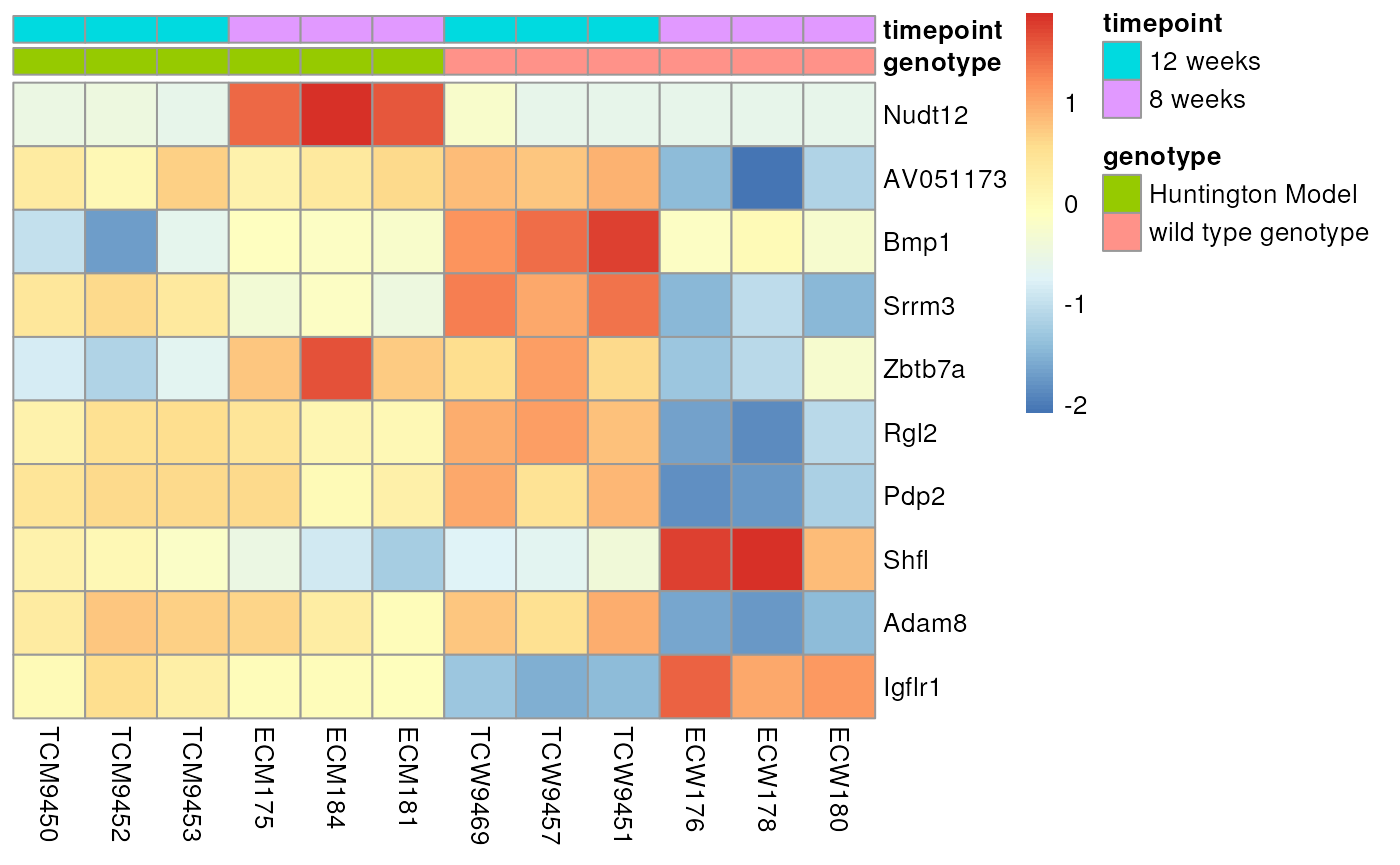
Session info
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] purrr_1.2.2 pheatmap_1.0.13 dplyr_1.2.1 gemma.R_3.9.2
## [5] BiocStyle_2.40.0
##
## loaded via a namespace (and not attached):
## [1] rappdirs_0.3.4 sass_0.4.10 generics_0.1.4
## [4] xml2_1.5.2 stringi_1.8.7 digest_0.6.39
## [7] magrittr_2.0.5 timechange_0.4.0 evaluate_1.0.5
## [10] grid_4.6.0 RColorBrewer_1.1-3 bookdown_0.47
## [13] fastmap_1.2.0 jsonlite_2.0.0 BiocManager_1.30.27
## [16] httr_1.4.8 viridisLite_0.4.3 scales_1.4.0
## [19] textshaping_1.0.5 jquerylib_0.1.4 cli_3.6.6
## [22] rlang_1.2.0 bit64_4.8.2 withr_3.0.2
## [25] cachem_1.1.0 yaml_2.3.12 otel_0.2.0
## [28] tools_4.6.0 memoise_2.0.1 kableExtra_1.4.0
## [31] assertthat_0.2.1 curl_7.1.0 vctrs_0.7.3
## [34] R6_2.6.1 lubridate_1.9.5 lifecycle_1.0.5
## [37] stringr_1.6.0 fs_2.1.0 htmlwidgets_1.6.4
## [40] bit_4.6.0 ragg_1.5.2 pkgconfig_2.0.3
## [43] desc_1.4.3 pkgdown_2.2.0 pillar_1.11.1
## [46] bslib_0.11.0 gtable_0.3.6 data.table_1.18.4
## [49] glue_1.8.1 systemfonts_1.3.2 xfun_0.58
## [52] tibble_3.3.1 tidyselect_1.2.1 rstudioapi_0.19.0
## [55] knitr_1.51 farver_2.1.2 htmltools_0.5.9
## [58] svglite_2.2.2 rmarkdown_2.31 compiler_4.6.0