Accessing curated gene expression data with gemma.R
Javier Castillo-Arnemann
Michael Smith Laboratories, University of British Columbia, Vancouver, CanadaJordan Sicherman
Michael Smith Laboratories, University of British Columbia, Vancouver, CanadaB. Ogan Mancarci
Michael Smith Laboratories, University of British Columbia, Vancouver, CanadaSource:
vignettes/gemma.R.Rmd
gemma.R.Rmd
library(gemma.R)
library(data.table)
library(dplyr)
library(ggplot2)
library(ggrepel)
library(SummarizedExperiment)
library(pheatmap)
library(viridis)
library(listviewer)About Gemma
Gemma is a web site, database and a set of tools for the meta-analysis, re-use and sharing of genomics data, currently primarily targeted at the analysis of gene expression profiles. Gemma contains data from thousands of public studies, referencing thousands of published papers. Every dataset in Gemma has passed a rigorous curation process that re-annotates the expression platform at the sequence level, which allows for more consistent cross-platform comparisons and meta-analyses.
For detailed information on the curation process, read this page or the latest publication.
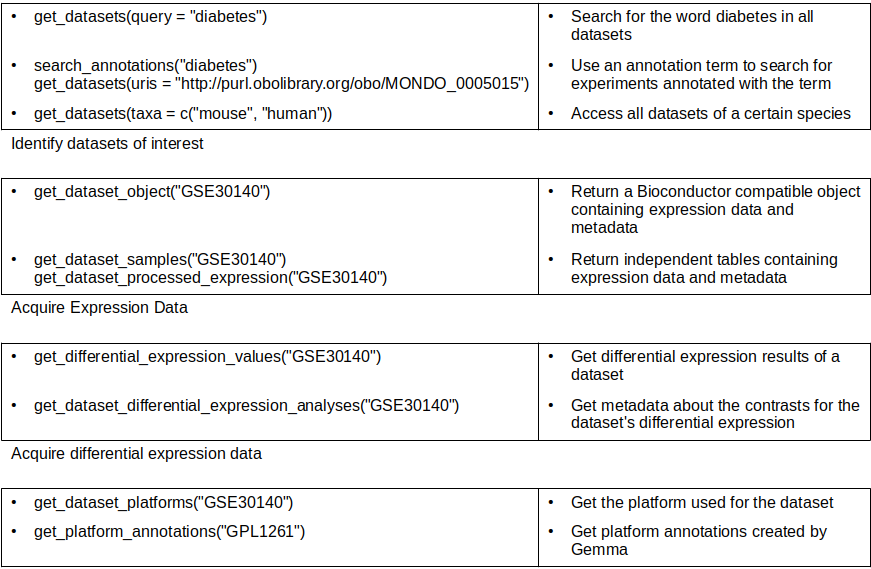
Installation instructions
Bioconductor
You can install gemma.R through Bioconductor with the following
code:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("gemma.R")Searching for datasets of interest in Gemma
Using the get_datasets
function, datasets fitting various criteria can be accessed.
# accessing all mouse and human datasets
get_datasets(taxa = c('mouse','human')) %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE2018 | Human Lung Transplant - BAL | Bronchoalveolar lavage samp… | human |
| GSE4523 | Expression Studies of Melan… | Melanotransferrin (MTf) or … | mouse |
| GSE4036 | perro-affy-human-186940 | Our laboratory has develope… | human |
| GSE4034 | palme-affy-mouse-198967 | Fear conditioning (FC) is a… | mouse |
| GSE2866 | Donarum-3R01NS040270-03S1 | Succinate semialdehyde dehy… | mouse |
| GSE3253 | Exaggerated neuroinflammati… | Acute cognitive impairment … | mouse |
# accessing human datasets with the word "bipolar"
get_datasets(query = 'bipolar',taxa = 'human') %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE5389 | Adult postmortem brain tiss… | Bipolar affective disorder … | human |
| GSE4030 | bunge-affy-arabi-162779 | Schwann cells, expanded in … | human |
| GSE5388 | Adult postmortem brain tiss… | Bipolar affective disorder … | human |
| GSE7036 | Expression profiling in mon… | To identify genes dysregula… | human |
| McLean Hippocampus | McLean Hippocampus | Hippocampus of schizophreni… | human |
| McLean_PFC | McLean_PFC | Prefrontal cortex of schizo… | human |
# access human datasets that were annotated with the ontology term for the
# bipolar disorder
# use search_annotations function to search for available annotation terms
get_datasets(taxa ='human',
uris = 'http://purl.obolibrary.org/obo/MONDO_0004985') %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE5389 | Adult postmortem brain tiss… | Bipolar affective disorder … | human |
| GSE5388 | Adult postmortem brain tiss… | Bipolar affective disorder … | human |
| GSE7036 | Expression profiling in mon… | To identify genes dysregula… | human |
| McLean Hippocampus | McLean Hippocampus | Hippocampus of schizophreni… | human |
| McLean_PFC | McLean_PFC | Prefrontal cortex of schizo… | human |
| stanley_feinberg | Stanley consortium collecti… | 50 samples of individuals f… | human |
get_dataset function also includes a filter
parameter that allows filtering for datasets with specific properties in
a more structured manner. A list of the available properties can be
accessed using filter_properties
filter_properties()$dataset %>% head %>% gemma_kable()| properties | type | description |
|---|---|---|
| accession.accession | string | NA |
| accession.accessionVersion | string | NA |
| accession.externalDatabase…. | string | NA |
| accession.externalDatabase.id | integer | NA |
| accession.externalDatabase…. | string | NA |
| accession.externalDatabase…. | string | NA |
These properties can be used together to fine tune your results
# access human datasets that has bipolar disorder as an experimental factor
get_datasets(taxa = 'human',
filter = "experimentalDesign.experimentalFactors.factorValues.characteristics.valueUri = http://purl.obolibrary.org/obo/MONDO_0004985") %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE5389 | Adult postmortem brain tiss… | Bipolar affective disorder … | human |
| GSE5388 | Adult postmortem brain tiss… | Bipolar affective disorder … | human |
| GSE7036 | Expression profiling in mon… | To identify genes dysregula… | human |
| McLean_PFC | McLean_PFC | Prefrontal cortex of schizo… | human |
| stanley_feinberg | Stanley consortium collecti… | 50 samples of individuals f… | human |
| stanley_kato | Stanley array collection DL… | 102 samples of individuals … | human |
# all datasets with more than 4 samples annotated for any disease
get_datasets(filter = 'bioAssayCount > 4 and allCharacteristics.category = disease') %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE2018 | Human Lung Transplant - BAL | Bronchoalveolar lavage samp… | human |
| GSE4036 | perro-affy-human-186940 | Our laboratory has develope… | human |
| GSE2866 | Donarum-3R01NS040270-03S1 | Succinate semialdehyde dehy… | mouse |
| GSE2426 | Pre-Neoplastic Stage of Med… | SUMMARY Medulloblastoma is… | mouse |
| GSE2867 | Zoghbi-5R01NS027699-14 | A number of human neurodege… | mouse |
| GSE3489 | Patterns of gene dysregulat… | The neurodegenerative proce… | human |
# all datasets with ontology terms for Alzheimer's disease and Parkinson's disease
# this is equivalent to using the uris parameter
get_datasets(filter = 'allCharacteristics.valueUri in (http://purl.obolibrary.org/obo/MONDO_0004975,http://purl.obolibrary.org/obo/MONDO_0005180
)') %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE4757 | Rogers-3U24NS043571-01S1 | Alzheimer’s Disease (AD) is… | human |
| GSE6613 | Parkinson’s disease vs. con… | Parkinson?s disease (PD) pr… | human |
| GSE12685 | Expression of mRNAs Regulat… | In Alzheimer’s disease (AD)… | human |
| GSE14499 | Effect of BDNF on the APP t… | We examined transgenic (TG)… | mouse |
| GSE10908 | Differential gene expressio… | In a transgenic mouse model… | mouse |
| GSE1297 | Incipient Alzheimer’s Disea… | For these data, we analyzed… | human |
Note that a single call of these functions will only return 20
results by default and a 100 results maximum, controlled by the
limit argument. In order to get all available results, use
get_all_pages function on the output of the function
get_datasets(taxa = 'human') %>%
get_all_pages() %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE2018 | Human Lung Transplant - BAL | Bronchoalveolar lavage samp… | human |
| GSE4036 | perro-affy-human-186940 | Our laboratory has develope… | human |
| GSE3489 | Patterns of gene dysregulat… | The neurodegenerative proce… | human |
| GSE1923 | Identification of PDGF-depe… | Overall study: Identificati… | human |
| GSE361 | Mammary epithelial cell tra… | Analysis of gene expression… | human |
| GSE492 | Effect of prostaglandin ana… | The purpose of this study i… | human |
See larger queries section for more details. To keep this vignette simpler we will keep using the first 20 results returned by default in examples below.
Dataset information provided by get_datasets also
includes some quality information that can be used to determine the
suitability of any given experiment. For instance
experiment.batchEffect column will be set to -1 if Gemma’s
preprocessing has detected batch effects that were unable to be resolved
by batch correction. More information about these and other fields can
be found at the function documentation.
get_datasets(taxa = 'human', filter = 'bioAssayCount > 4') %>%
filter(experiment.batchEffect !=-1) %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE2018 | Human Lung Transplant - BAL | Bronchoalveolar lavage samp… | human |
| GSE4036 | perro-affy-human-186940 | Our laboratory has develope… | human |
| GSE3489 | Patterns of gene dysregulat… | The neurodegenerative proce… | human |
| GSE1923 | Identification of PDGF-depe… | Overall study: Identificati… | human |
| GSE361 | Mammary epithelial cell tra… | Analysis of gene expression… | human |
| GSE492 | Effect of prostaglandin ana… | The purpose of this study i… | human |
Gemma uses multiple ontologies when annotating datasets and using the
term URIs instead of free text to search can lead to more specific
results. search_annotations
function allows searching for annotation terms that might be relevant to
your query.
search_annotations('bipolar') %>%
head %>% gemma_kable()| category.name | category.URI | value.name | value.URI |
|---|---|---|---|
| NA | NA | bipolar i disorder | http://www.ebi…/EFO_0009963 |
| NA | NA | bipolar ii disorder | http://www.ebi…/EFO_0009964 |
| NA | NA | bipolar affective disorder | http://purl.obolibrary…/HP_0007302 |
| NA | NA | bipolar i disorder | http://purl.obolibrary…/MONDO_0001866 |
| NA | NA | bipolar ii disorder | http://purl.obolibrary…/MONDO_0000693 |
| NA | NA | bipolar disorder | http://purl.obolibrary…/MONDO_0004985 |
Downloading expression data
Upon identifying datasets of interest, more information about specific ones can be requested. In this example we will be using GSE46416 which includes samples taken from healthy donors along with manic/euthymic phase bipolar disorder patients.
The data associated with specific experiments can be accessed by
using get_datasets_by_ids
get_datasets_by_ids("GSE46416") %>%
select(experiment.shortName, experiment.name,
experiment.description,taxon.name) %>%
head %>% gemma_kable| experiment.shortName | experiment.name | experiment.description | taxon.name |
|---|---|---|---|
| GSE46416 | State- and trait-specific g… | Gene expression profiles of… | human |
To access the expression data in a convenient form, you can use get_dataset_object.
It is a high-level wrapper that combines various endpoint calls to
return lists of annotated SummarizedExperiment
or ExpressionSet
objects that are compatible with other Bioconductor packages or a tidyverse-friendly
long form tibble for downstream analyses. These include the expression
matrix along with the experimental design, and ensure the sample names
match between both when transforming/subsetting data.
dat <- get_dataset_object("GSE46416",
type = 'se') # SummarizedExperiment is the default output typeNote that the tidy format is less memory efficient but allows easy visualization and exploration with ggplot2 and the rest of the tidyverse.
To show how subsetting works, we’ll keep the “manic phase” data and
the reference_subject_roles, which refers to the control
samples in Gemma datasets.
[1] "bipolar disorder with euthymic phase"
[2] "reference subject role"
[3] "bipolar disorder with manic phase"
# Subset patients during manic phase and controls
manic <- dat[[1]][, dat[[1]]$disease == "bipolar disorder with manic phase" |
dat[[1]]$disease == "reference subject role"]
manicclass: SummarizedExperiment
dim: 18758 21
metadata(9): title abstract ... gemmaSuitabilityScore taxon
assays(1): counts
rownames(18758): 2315430 2315554 ... 7385683 7385696
rowData names(4): Probe GeneSymbol GeneName NCBIid
colnames(21): Control, 12 Control, 1_DE50 ... Bipolar disorder patient
manic phase, 21 Bipolar disorder patient manic phase, 16
colData names(3): factorValues block diseaseLet’s take a look at sample to sample correlation in our subset.
# Get Expression matrix
manicExpr <- assay(manic, "counts")
manicExpr %>%
cor %>%
pheatmap(col =viridis(10),border_color = NA,angle_col = 45,fontsize = 7)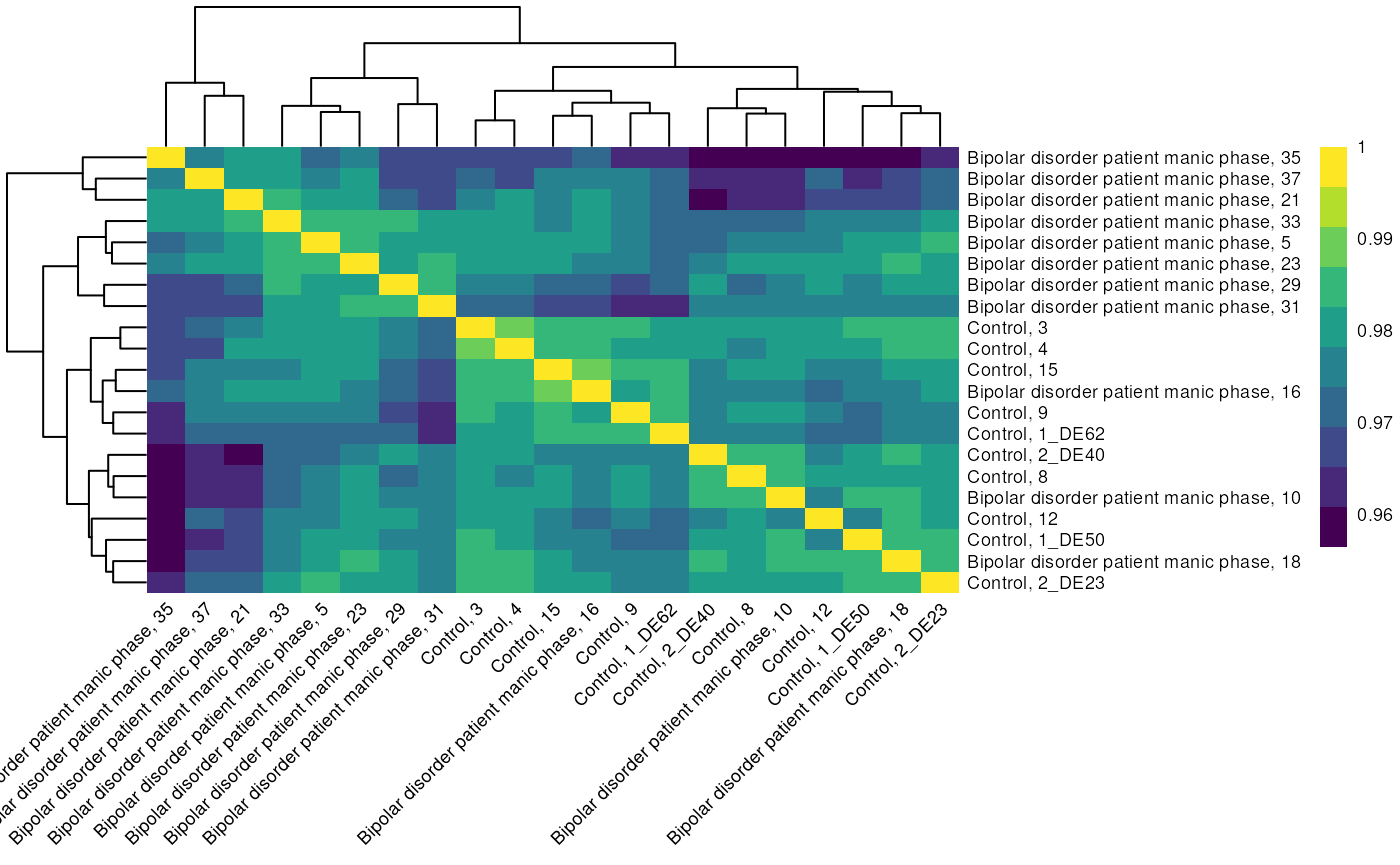
Sample to sample correlations of bipolar patients during manic phase and controls.
You can also use get_dataset_processed_expression
to only get the expression matrix, get_dataset_samples
to get the metadata information. The output of this function includes
some additional details about a sample such as the original accession ID
or whether or not it was determined to be an outlier but it can be
simplified to match the design table included in the output of
get_dataset_object by using make_design
on the output.
get_dataset_samples('GSE46416') %>% make_design('text') %>% select(-factorValues) %>% head %>%
gemma_kable()| block | disease | |
|---|---|---|
| Bipolar disorder patient euthymic phase, 11 | Batch_02_26/11/09 | bipolar disorder with euthy… |
| Bipolar disorder patient euthymic phase, 17 | Batch_02_26/11/09 | bipolar disorder with euthy… |
| Control, 12 | Batch_02_26/11/09 | reference subject role |
| Control, 1_DE50 | Batch_05_24/11/10 | reference subject role |
| Bipolar disorder patient euthymic phase, 19 | Batch_03_27/11/09 | bipolar disorder with euthy… |
| Control, 9 | Batch_01_25/11/09 | reference subject role |
Platform Annotations
Expression data in Gemma comes with annotations for the gene each
expression profile corresponds to. Using the get_platform_annotations
function, these annotations can be retrieved independently of the
expression data, along with additional annotations such as Gene Ontology
terms.
Examples:
head(get_platform_annotations('GPL96') %>% select(-GOTerms)) ElementName GeneSymbols
<char> <char>
1: 202619_s_at PLOD2
2: 219952_s_at MCOLN1
3: 205192_at MAP3K14
4: 212724_at RND3
5: 207373_at HOXD10
6: 204991_s_at NF2
GeneNames GemmaIDs NCBIids
<char> <char> <char>
1: procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 127276 5352
2: mucolipin TRP cation channel 1 313978 57192
3: mitogen-activated protein kinase kinase kinase 14 188876 9020
4: Rho family GTPase 3 9353 390
5: homeobox D10 72301 3236
6: NF2, moesin-ezrin-radixin like (MERLIN) tumor suppressor 116071 4771
EnsemblIds
<char>
1: ENSG00000152952
2: ENSG00000090674
3: ENSG00000006062
4: ENSG00000115963
5: ENSG00000128710
6: ENSG00000186575
head(get_platform_annotations('Generic_human_ncbiIds') %>% select(-GOTerms)) ElementName GeneSymbols GeneNames GemmaIDs
<int> <char> <char> <int>
1: 105373804 LOC105373804 uncharacterized LOC105373804 9236458
2: 285540 SEPSECS-AS1 SEPSECS antisense RNA 1 (head to head) 421333
3: 105373805 STAT4-AS1 STAT4 antisense RNA 1 9236463
4: 107985219 LOC107985219 uncharacterized LOC107985219 9667149
5: 105373802 NA
6: 107985216 LOC107985216 uncharacterized LOC107985216 9667148
NCBIids EnsemblIds
<int> <char>
1: 105373804
2: 285540
3: 105373805 ENSG00000231858
4: 107985219
5: NA
6: 107985216 If you are interested in a particular gene, you can see which
platforms include it using get_gene_probes.
Note that functions to search gene work best with unambigious
identifiers rather than symbols.
# lists genes in gemma matching the symbol or identifier
get_genes('Eno2') %>% gemma_kable()| gene.symbol | gene.ensembl | gene.NCBI | gene.name | gene.aliases | gene.MFX.rank | taxon.name | taxon.scientific | taxon.ID | taxon.NCBI | taxon.database.name | taxon.database.ID |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ENO2 | ENSG00000111674 | 2026 | enolase 2 | HEL-S-27…. | 0.8776645 | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| Eno2 | ENSMUSG00000004267 | 13807 | enolase 2, gamma neuronal | D6Ertd37…. | 0.8794051 | mouse | Mus musculus | 2 | 10090 | mm10 | 81 |
| Eno2 | ENSRNOG00000013141 | 24334 | enolase 2 | NSE, RNEN3 | 0.8187614 | rat | Rattus norvegicus | 3 | 10116 | rn6 | 86 |
# ncbi id for human ENO2
probes <- get_gene_probes(2026)
# remove the description for brevity of output
head(probes[,.SD, .SDcols = !colnames(probes) %in% c('mapping.Description','platform.Description')]) %>%
gemma_kable()| element.name | element.description | platform.shortName | platform.name | platform.ID | platform.type | platform.description | platform.troubled | taxon.name | taxon.scientific | taxon.ID | taxon.NCBI | taxon.database.name | taxon.database.ID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 201313_at | enolase 2 (gamma, neuronal) | GPL96 | Affymetrix GeneChip Human G… | 1 | ONECOLOR | The U133 set includes 2 ar… | FALSE | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 201313_at | enolase 2 (gamma, neuronal) | GPL570 | Affymetrix GeneChip Human G… | 4 | ONECOLOR | Complete coverage of the H… | FALSE | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 40193_at | enolase 2 (gamma, neuronal) | GPL91 | Affymetrix GeneChip Human G… | 8 | ONECOLOR | The Human Genome U95 (HG-U… | FALSE | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 1639 | NA | GPL962 | CHUGAI 41K | 36 | TWOCOLOR | Patients and Sample Prepar… | FALSE | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 6621 | NA | GPL230 | UP_5Ka | 38 | TWOCOLOR | A cDNA microarray with ~50… | FALSE | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 861 | NA | GPL230 | UP_5Ka | 38 | TWOCOLOR | A cDNA microarray with ~50… | FALSE | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
Differential expression analyses
Gemma contains precomputed differential expression analyses for most
of its datasets. Analyses can involve more than one factor, such as
“sex” as well as “disease”. Some datasets contain more than one analysis
to account for different factors and their interactions. The results are
stored as resultSets, each corresponding to one factor (or their
interaction). You can access them using get_differential_expression_values.
From here on, we can explore and visualize the data to find the most
differentially-expressed genes
Note that get_differential_expression_values can return
multiple differentials per study if a study has multiple factors to
contrast. Since GSE46416 only has one extracting the first element of
the returned list is all we need.
dif_exp <- get_differential_expression_values('GSE46416')
dif_exp[[1]] %>% head %>% gemma_kable()| Probe | NCBIid | gene_ensembl_id | GeneSymbol | GeneName | pvalue | corrected_pvalue | rank | contrast_113004_coefficient | contrast_113004_log2fc | contrast_113004_tstat | contrast_113004_pvalue | contrast_113005_coefficient | contrast_113005_log2fc | contrast_113005_tstat | contrast_113005_pvalue |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3563604 | 283551 | ENSG00000214900 | LINC01588 | long intergenic non-protein… | 9.0e-07 | 0.0028 | 3.00e-04 | -0.5543 | -0.5543 | -5.7640 | 2.10e-06 | -0.5617 | -0.5617 | -5.8407 | 1.7e-06 |
| 3361181 | 387751 | ENSG00000254838 | GVINP1 | GTPase, very large interfer… | 9.0e-07 | 0.0028 | 2.00e-04 | -0.5702 | -0.5702 | -4.0491 | 3.00e-04 | -0.9326 | -0.9326 | -6.6228 | 2.0e-07 |
| 2362351 | 149628 | ENSG00000163564 | PYHIN1 | pyrin and HIN domain family… | 5.0e-07 | 0.0028 | 1.00e-04 | -0.7474 | -0.7474 | -5.3795 | 6.40e-06 | -0.8938 | -0.8938 | -6.4326 | 3.0e-07 |
| 3328214 | 221120 | ENSG00000166199 | ALKBH3 | alkB homolog 3, alpha-ketog… | 4.0e-07 | 0.0028 | 9.11e-05 | -0.2252 | -0.2252 | -4.0499 | 3.00e-04 | -0.3849 | -0.3849 | -6.9210 | 1.0e-07 |
| 3918696 | 6651 | ENSG00000159140 | SON | SON DNA and RNA binding pro… | 6.0e-07 | 0.0028 | 2.00e-04 | -0.2222 | -0.2222 | -4.1695 | 2.00e-04 | -0.3588 | -0.3588 | -6.7310 | 1.0e-07 |
| 3406015 | 55729 | ENSG00000171681 | ATF7IP | activating transcription fa… | 1.1e-06 | 0.0031 | 4.00e-04 | -0.3672 | -0.3672 | -4.5202 | 7.86e-05 | -0.5220 | -0.5220 | -6.4259 | 3.0e-07 |
By default the columns names of the output correspond to contrast
IDs. To see what conditions these IDs correspond to we can either use
get_dataset_differential_expression_analyses to get the
metadata about differentials of a given dataset, or set
readableContrasts argument of
get_differential_expression_values to TRUE.
The former approach is usually better for a large scale systematic
analysis while the latter is easier to read in an interactive
session.
get_dataset_differential_expression_analyses function
returns structured metadata about the differentials.
contrasts <- get_dataset_differential_expression_analyses('GSE46416')
contrasts %>% gemma_kable()| result.ID | contrast.ID | experiment.ID | factor.category | factor.category.URI | factor.ID | baseline.factors | experimental.factors | isSubset | subsetFactor | probes.analyzed | genes.analyzed |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 550248 | 113004 | 8997 | disease | http://www.ebi…/EFO_0000408 | 19134 | disease,…. | disease,…. | FALSE | logical(…. | 21961 | 18959 |
| 550248 | 113005 | 8997 | disease | http://www.ebi…/EFO_0000408 | 19134 | disease,…. | disease,…. | FALSE | logical(…. | 21961 | 18959 |
contrast.ID column corresponds to the column names in
the output of get_differential_expression_values while
result.ID corresponds to the name of the differential in
the output object. Using them together will let one to access
differentially expressed gene counts for each condition contrast
# using result.ID and contrast.ID of the output above, we can access specific
# results. Note that one study may have multiple contrast objects
seq_len(nrow(contrasts)) %>% sapply(function(i){
result_set = dif_exp[[as.character(contrasts[i,]$result.ID)]]
p_values = result_set[[glue::glue("contrast_{contrasts[i,]$contrast.ID}_pvalue")]]
# multiple testing correction
sum(p.adjust(p_values,method = 'BH') < 0.05)
}) -> dif_exp_genes
contrasts <- data.table(result.ID = contrasts$result.ID,
contrast.id = contrasts$contrast.ID,
baseline.factorValue = contrasts$baseline.factors,
experimental.factorValue = contrasts$experimental.factors,
n_diff = dif_exp_genes)
contrasts %>% gemma_kable()| result.ID | contrast.id | baseline.factorValue | experimental.factorValue | n_diff |
|---|---|---|---|---|
| 550248 | 113004 | disease,…. | disease,…. | 1 |
| 550248 | 113005 | disease,…. | disease,…. | 1294 |
contrasts$baseline.factorsNULL
contrasts$experimental.factorsNULLAlternatively we, since we are only looking at one dataset and one
contrast manually, we can simply use readableContrasts.
de <- get_differential_expression_values("GSE46416",readableContrasts = TRUE)[[1]]
de %>% head %>% gemma_kable| Probe | NCBIid | gene_ensembl_id | GeneSymbol | GeneName | pvalue | corrected_pvalue | rank | contrast_bipolar disorder with manic phase_coefficient | contrast_bipolar disorder with manic phase_logFoldChange | contrast_bipolar disorder with manic phase_tstat | contrast_bipolar disorder with manic phase_pvalue | contrast_bipolar disorder with euthymic phase_coefficient | contrast_bipolar disorder with euthymic phase_logFoldChange | contrast_bipolar disorder with euthymic phase_tstat | contrast_bipolar disorder with euthymic phase_pvalue |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3563604 | 283551 | ENSG00000214900 | LINC01588 | long intergenic non-protein… | 9.0e-07 | 0.0028 | 3.00e-04 | -0.5543 | -0.5543 | -5.7640 | 2.10e-06 | -0.5617 | -0.5617 | -5.8407 | 1.7e-06 |
| 3361181 | 387751 | ENSG00000254838 | GVINP1 | GTPase, very large interfer… | 9.0e-07 | 0.0028 | 2.00e-04 | -0.5702 | -0.5702 | -4.0491 | 3.00e-04 | -0.9326 | -0.9326 | -6.6228 | 2.0e-07 |
| 2362351 | 149628 | ENSG00000163564 | PYHIN1 | pyrin and HIN domain family… | 5.0e-07 | 0.0028 | 1.00e-04 | -0.7474 | -0.7474 | -5.3795 | 6.40e-06 | -0.8938 | -0.8938 | -6.4326 | 3.0e-07 |
| 3328214 | 221120 | ENSG00000166199 | ALKBH3 | alkB homolog 3, alpha-ketog… | 4.0e-07 | 0.0028 | 9.11e-05 | -0.2252 | -0.2252 | -4.0499 | 3.00e-04 | -0.3849 | -0.3849 | -6.9210 | 1.0e-07 |
| 3918696 | 6651 | ENSG00000159140 | SON | SON DNA and RNA binding pro… | 6.0e-07 | 0.0028 | 2.00e-04 | -0.2222 | -0.2222 | -4.1695 | 2.00e-04 | -0.3588 | -0.3588 | -6.7310 | 1.0e-07 |
| 3406015 | 55729 | ENSG00000171681 | ATF7IP | activating transcription fa… | 1.1e-06 | 0.0031 | 4.00e-04 | -0.3672 | -0.3672 | -4.5202 | 7.86e-05 | -0.5220 | -0.5220 | -6.4259 | 3.0e-07 |
# Classify probes for plotting
de$diffexpr <- "No"
de$diffexpr[de$`contrast_bipolar disorder with manic phase_logFoldChange` > 1.0 &
de$`contrast_bipolar disorder with manic phase_pvalue` < 0.05] <- "Up"
de$diffexpr[de$`contrast_bipolar disorder with manic phase_logFoldChange` < -1.0 &
de$`contrast_bipolar disorder with manic phase_pvalue` < 0.05] <- "Down"
# Upregulated probes
filter(de, diffexpr == "Up") %>%
arrange(`contrast_bipolar disorder with manic phase_pvalue`) %>%
select(Probe, GeneSymbol, `contrast_bipolar disorder with manic phase_pvalue`,
`contrast_bipolar disorder with manic phase_logFoldChange`) %>%
head(10) %>% gemma_kable()| Probe | GeneSymbol | contrast_bipolar disorder with manic phase_pvalue | contrast_bipolar disorder with manic phase_logFoldChange |
|---|---|---|---|
| 2319550 | RBP7 | 8.61e-05 | 1.0740 |
| 2548699 | CYP1B1 | 1.00e-04 | 1.3225 |
| 3907190 | SLPI | 3.00e-04 | 1.0558 |
| 3629103 | PCLAF | 5.00e-04 | 1.2783 |
| 3545525 | SLIRP | 6.00e-04 | 1.3490 |
| 3146433 | COX6C | 9.00e-04 | 1.4670 |
| 2899102 | H3C3 | 1.30e-03 | 1.0260 |
| 3635198 | BCL2A1 | 1.80e-03 | 1.0798 |
| 2633191 | GPR15 | 2.40e-03 | 1.2046 |
| 3518169 | COMMD6 | 2.50e-03 | 1.3763 |
# Downregulated probes
filter(de, diffexpr == "Down") %>%
arrange(`contrast_bipolar disorder with manic phase_pvalue`) %>%
select(Probe, GeneSymbol, `contrast_bipolar disorder with manic phase_pvalue`,
`contrast_bipolar disorder with manic phase_logFoldChange`) %>%
head(10) %>% gemma_kable()| Probe | GeneSymbol | contrast_bipolar disorder with manic phase_pvalue | contrast_bipolar disorder with manic phase_logFoldChange |
|---|---|---|---|
| 3245871 | WDFY4 | 0.0002 | -1.1569 |
| 3022689 | SND1-IT1 | 0.0002 | -1.2199 |
| 3384417 | ANKRD42-DT | 0.0004 | -1.0030 |
| 3930525 | RUNX1-IT1 | 0.0004 | -1.5169 |
| 3336402 | RBM14 | 0.0004 | -1.0711 |
| 3652609 | SMG1P2 | 0.0005 | -1.2544 |
| 2663083 | TAMM41 | 0.0005 | -1.3056 |
| 3404030 | KLRG1 | 0.0007 | -1.0949 |
| 3041550 | TRA2A | 0.0008 | -1.0496 |
| 3526425 | PCID2 | 0.0011 | -1.0719 |
# Add gene symbols as labels to DE genes
de$delabel <- ""
de$delabel[de$diffexpr != "No"] <- de$GeneSymbol[de$diffexpr != "No"]
# Volcano plot for bipolar patients vs controls
ggplot(
data = de,
aes(
x = `contrast_bipolar disorder with manic phase_logFoldChange`,
y = -log10(`contrast_bipolar disorder with manic phase_pvalue`),
color = diffexpr,
label = delabel
)
) +
geom_point() +
geom_hline(yintercept = -log10(0.05), col = "gray45", linetype = "dashed") +
geom_vline(xintercept = c(-1.0, 1.0), col = "gray45", linetype = "dashed") +
labs(x = "log2(FoldChange)", y = "-log10(p-value)") +
scale_color_manual(values = c("blue", "black", "red")) +
geom_text_repel(show.legend = FALSE) +
theme_minimal()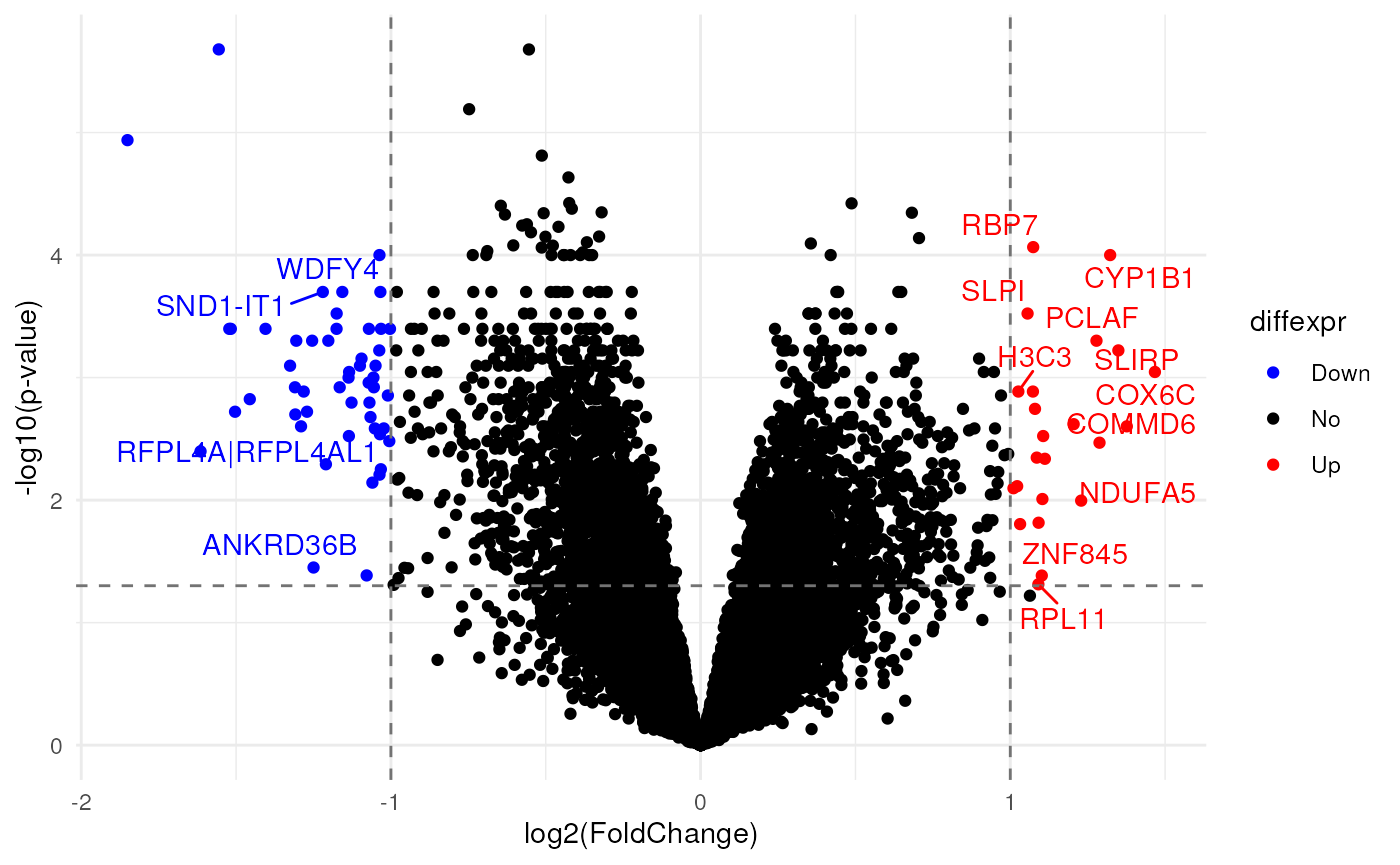
Differentially-expressed genes in bipolar patients during manic phase versus controls.
Larger queries
To query large amounts of data, the API has a pagination system which
uses the limit and offset parameters. To avoid
overloading the server, calls are limited to a maximum of 100 entries,
so the offset allows you to get the next batch of entries in the next
call(s).
To simplify the process of accessing all available data, gemma.R
includes the get_all_pages
function which can use the output from one page to make all the follow
up requests
get_platforms_by_ids() %>%
get_all_pages() %>% head %>% gemma_kable()| platform.ID | platform.shortName | platform.name | platform.description | platform.troubled | platform.experimentCount | platform.type | taxon.name | taxon.scientific | taxon.ID | taxon.NCBI | taxon.database.name | taxon.database.ID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | GPL96 | Affymetrix GeneChip Human G… | The U133 set includes 2 ar… | FALSE | 397 | ONECOLOR | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 2 | GPL1355 | Affymetrix GeneChip Rat Gen… | The GeneChip Rat Genome 23… | FALSE | 297 | ONECOLOR | rat | Rattus norvegicus | 3 | 10116 | rn6 | 86 |
| 3 | GPL1261 | Affymetrix GeneChip Mouse G… | All probe sets represented… | FALSE | 1325 | ONECOLOR | mouse | Mus musculus | 2 | 10090 | mm10 | 81 |
| 4 | GPL570 | Affymetrix GeneChip Human G… | Complete coverage of the H… | FALSE | 1562 | ONECOLOR | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 5 | GPL81 | Affymetrix GeneChip Murine … | The MG-U74 set includes 3 … | FALSE | 188 | ONECOLOR | mouse | Mus musculus | 2 | 10090 | mm10 | 81 |
| 6 | GPL85 | Affymetrix GeneChip Rat Gen… | The RG-U34 set includes 3 … | FALSE | 84 | ONECOLOR | rat | Rattus norvegicus | 3 | 10116 | rn6 | 86 |
Alternative way to access all pages is to do so manually. To see how many available results are there, you can look at the attributes of the output objects where additional information from the API response is appended.
platform_count = attributes(get_platforms_by_ids(limit = 1))$totalElements
print(platform_count)[1] 670After which you can use offset to access all available
platforms.
lapply(seq(0,platform_count,100), function(offset){
get_platforms_by_ids(limit = 100, offset = offset) %>%
select(platform.ID, platform.shortName, taxon.name)
}) %>% do.call(rbind,.) %>%
head %>% gemma_kable()| platform.ID | platform.shortName | taxon.name |
|---|---|---|
| 1 | GPL96 | human |
| 2 | GPL1355 | rat |
| 3 | GPL1261 | mouse |
| 4 | GPL570 | human |
| 5 | GPL81 | mouse |
| 6 | GPL85 | rat |
Many endpoints only support a single identifier:
get_dataset_annotations(c("GSE35974", "GSE46416"))
[1m
[33mError
[39m:
[22m
[33m!
[39m Please specify one valid identifier for dataset.In these cases, you will have to loop over all the identifiers you wish to query and send separate requests.
lapply(c("GSE35974", "GSE12649"), function(dataset) {
get_dataset_annotations(dataset) %>%
mutate(experiment.shortName = dataset) %>%
select(experiment.shortName, class.name, term.name)
}) %>% do.call(rbind,.) %>% gemma_kable()| experiment.shortName | class.name | term.name |
|---|---|---|
| GSE35974 | organism part | cerebellum |
| GSE35974 | assay | transcription profiling by … |
| GSE35974 | disease | schizophrenia |
| GSE35974 | disease | bipolar disorder |
| GSE35974 | disease | depressive disorder |
| GSE35974 | biological sex | male |
| GSE35974 | labelling | biotin |
| GSE12649 | organism part | prefrontal cortex |
| GSE12649 | assay | transcription profiling by … |
| GSE12649 | disease | schizophrenia |
| GSE12649 | disease | bipolar disorder |
| GSE12649 | labelling | biotin |
Output options
Raw data
By default, Gemma API does some parsing on the raw API results to
make it easier to work with inside of R. In the process, it drops some
typically unused values. If you wish to fetch everything, use
raw = TRUE. Instead of a data table, you’ll usually be
served a list that represents the underlying JSON response.
get_gene_locations("DYRK1A") %>% gemma_kable()| chromosome | strand | nucleotide | length | taxon.name | taxon.scientific | taxon.ID | taxon.NCBI | taxon.database.name | taxon.database.ID |
|---|---|---|---|---|---|---|---|---|---|
| 11 |
|
33890705 | 118714 | rat | Rattus norvegicus | 3 | 10116 | rn6 | 86 |
| 21 |
|
37365572 | 160785 | human | Homo sapiens | 1 | 9606 | hg38 | 87 |
| 16 |
|
94370769 | 125608 | mouse | Mus musculus | 2 | 10090 | mm10 | 81 |
get_gene_locations("DYRK1A", raw = TRUE) %>% jsonedit()File outputs
Sometimes, you may wish to save results to a file for future
inspection. You can do this simply by providing a filename to the
file parameter. The extension for this file will be one of
three options:
-
.json, if you requested results withraw=TRUE -
.csvif the results have no nested data tables -
.rdsotherwise
You can also specify whether or not the new fetched results are
allowed to overwrite an existing file by specifying the
overwrite = TRUE parameter.
Memoise data
To speed up results, you can remember past results so future queries
can proceed virtually instantly. This is enabled through the memoise
package. To enable memoisation, simply set memoised = TRUE
in the function call whenever you want to refer to the cache, both to
save data for future calls and use the saved data for repeated calls. By
default this will create a cache in your local filesystem.
If you wish to change where the cache is stored or change the default
behaviour to make sure you always use the cache without relying on the
memoised argument, use gemma_memoised.
# use memoisation by default using the default cache
gemma_memoised(TRUE)
# set an altnernate cache path
gemma_memoised(TRUE,"path/to/cache_directory")
# cache in memory of the R session
# this cache will not be preserved between sessions
gemma_memoised(TRUE,"cache_in_memory")If you’re done with your fetching and want to ensure no space is
being used for cached results, or if you just want to ensure you’re
getting up-to-date data from Gemma, you can clear the cache using forget_gemma_memoised.
Changing defaults
We’ve seen how to change raw = TRUE,
overwrite = TRUE and memoised = TRUE in
individual function calls. It’s possible that you want to always use the
functions these ways without specifying the option every time. You can
do this by simply changing the default, which is visible in the function
definition. See below for examples.
Session info
R version 4.6.0 (2026-04-24)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: UTC
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] listviewer_4.0.0 viridis_0.6.5
[3] viridisLite_0.4.3 pheatmap_1.0.13
[5] SummarizedExperiment_1.42.0 Biobase_2.72.0
[7] GenomicRanges_1.64.0 Seqinfo_1.2.0
[9] IRanges_2.46.0 S4Vectors_0.50.1
[11] BiocGenerics_0.58.1 generics_0.1.4
[13] MatrixGenerics_1.24.0 matrixStats_1.5.0
[15] ggrepel_0.9.8 ggplot2_4.0.3
[17] dplyr_1.2.1 data.table_1.18.4
[19] gemma.R_3.9.2 BiocStyle_2.40.0
loaded via a namespace (and not attached):
[1] tidyselect_1.2.1 farver_2.1.2 S7_0.2.2
[4] fastmap_1.2.0 digest_0.6.39 timechange_0.4.0
[7] lifecycle_1.0.5 magrittr_2.0.5 compiler_4.6.0
[10] rlang_1.2.0 sass_0.4.10 tools_4.6.0
[13] yaml_2.3.12 knitr_1.51 labeling_0.4.3
[16] S4Arrays_1.12.0 htmlwidgets_1.6.4 bit_4.6.0
[19] curl_7.1.0 DelayedArray_0.38.2 xml2_1.5.2
[22] RColorBrewer_1.1-3 abind_1.4-8 withr_3.0.2
[25] purrr_1.2.2 desc_1.4.3 grid_4.6.0
[28] scales_1.4.0 cli_3.6.6 rmarkdown_2.31
[31] ragg_1.5.2 otel_0.2.0 rstudioapi_0.19.0
[34] httr_1.4.8 cachem_1.1.0 stringr_1.6.0
[37] assertthat_0.2.1 BiocManager_1.30.27 XVector_0.52.0
[40] vctrs_0.7.3 Matrix_1.7-5 jsonlite_2.0.0
[43] bookdown_0.47 bit64_4.8.2 systemfonts_1.3.2
[46] jquerylib_0.1.4 glue_1.8.1 pkgdown_2.2.0
[49] lubridate_1.9.5 stringi_1.8.7 gtable_0.3.6
[52] tibble_3.3.1 pillar_1.11.1 rappdirs_0.3.4
[55] htmltools_0.5.9 R6_2.6.1 textshaping_1.0.5
[58] evaluate_1.0.5 kableExtra_1.4.0 lattice_0.22-9
[61] memoise_2.0.1 bslib_0.11.0 Rcpp_1.1.1-1.1
[64] svglite_2.2.2 gridExtra_2.3 SparseArray_1.12.2
[67] xfun_0.58 fs_2.1.0 pkgconfig_2.0.3